Gene Mutation of Congenital Lactase Deficiency (CLD)
Defects in lactase (LCT) gene are the cause of congenital lactase deficiency (also known as hereditary alactasia or disaccharide intolerance II). Congenital lactase deficiency is an autosomal recessive, rare and severe gastrointestinal disorder. It is characterized by watery diarrhea in infants fed with breast milk or other lactose-containing formulas. An almost total lack of LCT activity is found in jejunal biopsy material of patients with congenital lactase deficiency. Opposite to congenital lactase deficiency, adult-type hypolactasia, also known as lactose intolerance, is the most common enzyme deficiency worldwide. It is caused by developmental down-regulation of lactase activity during childhood or early adulthood (Kuokkanen et al., 2005). Symptoms can be avoided and patients can have normal growth and development by changing to a lactose-free diet (Torniainen et al., 2009). The decline of lactase activity is a normal physiological phenomenon; however, the majority of Northern Europeans have the ability to maintain lactase activity and digest lactose throughout life (lactase persistence). The down-regulation of lactase activity operates at the transcriptional level and it is associated with a non-coding variation in the MCM6 gene, located in the upstream vicinity of LCT (Kuokkanen et al., 2005).
Generally, there are 9 distinct mutations in the coding region of the lactase (LCT) gene:
Generally, there are 9 distinct mutations in the coding region of the lactase (LCT) gene:
- All the chromosomes with the major disease haplotype carried a nonsense mutation, c.4170TrA (Finmajor), resulting in Y1390X, a premature stop codon in exon 9 predicting the truncation of 537 amino acids. (Refer Table 1)
- A deletion of four nucleotides, c.4998_5001delTGAG, in their paternal disease chromosome in exon 14, leading to a frameshift and a premature stop codon after 55 altered amino acids (S1666fsX1722). (Refer Table 1)
- c.653_654delCT, is a deletion of two nucleotides in exon 2, predicting a frameshift change at codon 218 and protein truncation at codon 224, S218fsX224. (Refer Table 1)
- c.804GrC transversion at codon 268, leading to an amino acid substitution of histidine for glutamine, Q268H, in the last nucleotide of exon 3. (Refer Table 1)
- c.4087GrA transition resulting in a missense substitution of serine for an uncharged glycine, G1363S, at codon 1363 in exon 9. (Refer Table 1)
- Deletion of five bases c.1692-1696delAGTGG in exon 6 leading to frameshift mutation V565fsX567 was found in Finnish patient heterozygous for the founder mutation Y1390X. (Refer Figure 1)
- The c.2062T > C in exon 7 leading to a substitution of serine to proline. Substitution S688P is located in the region II of the pro-LPH that has been shown to have a role as an intramolecular chaperone in the folding of the LPH-protein. (Refer Figure 1)
- The c.4834G > T in exon 12 leading to glutamic acid to change to a premature stop codon. Mutations E1612X is located in the conserved region IV of the mature LCT that encodes lactase activity. Mutation E1612X leads to a truncated protein. (Refer Figure 1)
- Sequencing of the LCT gene resulted in the identification of substitution c.4760G→A in exon 12 which changes arginine to histidine. Mutations R1587H is located in the conserved region IV of the mature LCT that encodes lactase activity. In substitution R1587H both amino acids are polar, basic amino acids. However, arginine always has a positive charge and a role in the maintenance of protein overall charge balance. Therefore the substitution to histidine (which charge depends on environment) may disturb the overall charge. (Refer Figure 1)
Table 1 shows the Q268H residue is also implied by cross-species conservation from human through chimpanzee, rabbit, cow, rat, and mouse (Kuokkanen et al., 2005).
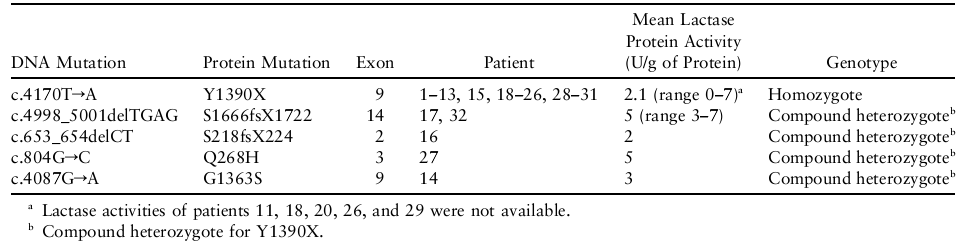
Table 1: CLD Mutations in the LCT Gene and Observed Lactase Activities of Duodenal Biopsy Specimens (Source: Kuokkanen. M et al. (2005). "Mutations in the Translated Region of the Lactase Gene (LCT) Underlie Congenital Lactase Deficiency." The American Journal of Human Genetics 78.)
Figure 1 shows the ClustalW alignment of human LPH protein and LPH proteins from other species showing the conservation of the region for mutations V565fsX567 (A), S688P (B), R1587H (C) and E1612X (C) (Torniainen et al., 2009).

Figure 1: Conservation of mutations sites (Source: Torniainen. S et al. (2009). "Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD)." BMC Gastroenterology.)
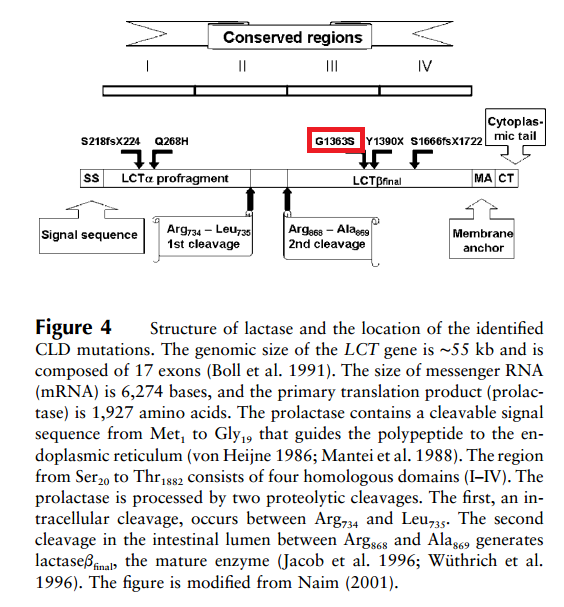
Figure 2: The schematic structure of lactase polypeptide and the location of the five CLD mutations (Source: Kuokkanen. M et al. (2005). "Mutations in the Translated Region of the Lactase Gene (LCT) Underlie Congenital Lactase Deficiency." The American Journal of Human Genetics 78.)
The fifth mutation, G1363S (as shown in the Figure 2 with red colour circle), replaces the smallest amino acid, glycine, with serine, which has a hydroxyl group that makes it polar and reactive. The replacement of the small glycine by the larger serine may alter the three-dimensional structure of the polypeptide. Since the G1363S mutation is located in the mature lactase at the end of region III, near the catalytic active sites, it may have serious functional consequences. Also, G1363 is conserved across species (Kuokkanen et al., 2005). Therefore, mutation at this point will cause the protein cannot be expressed and functioned properly.
In conclusion, this study confirms that both human lactase deficiencies are related to DNA variants affecting the LCT gene. Interestingly, the mutations resulting in the severe congenital form have direct consequences for the polypeptide leading to nonsense-mediated mRNA decay in at least 90% of the disease alleles, whereas the critical nucleotide for the milder adult-type phenotype represents a distal enhancer regulating the transcript levels in intestinal cells (Kuokkanen et al., 2005).
Sources:
In conclusion, this study confirms that both human lactase deficiencies are related to DNA variants affecting the LCT gene. Interestingly, the mutations resulting in the severe congenital form have direct consequences for the polypeptide leading to nonsense-mediated mRNA decay in at least 90% of the disease alleles, whereas the critical nucleotide for the milder adult-type phenotype represents a distal enhancer regulating the transcript levels in intestinal cells (Kuokkanen et al., 2005).
Sources:
- Kuokkanen. M et al. (2005). "Mutations in the Translated Region of the Lactase Gene (LCT) Underlie Congenital Lactase Deficiency." The American Journal of Human Genetics 78. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1380240/
- Torniainen. S et al. (2009). "Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD)." BMC Gastroenterology. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2635369/
